MS缺陷建模求助

xuekekekeke
各位大神,最近学习MS遇到好多问题,希望各位大佬不吝赐教!问题:插入缺陷前,用castep计算都正常,没有错误,建立2x2的超胞后,替换一部分原子,再进行晶格优化,就出现错误了,想请教各位大佬在进行缺陷建模时,一般是怎么做的,遇到我这种问题该怎么办。(求助)
第一原理
- 450
- 9
缺陷形成能VS费米能级图的疑问
2011932051
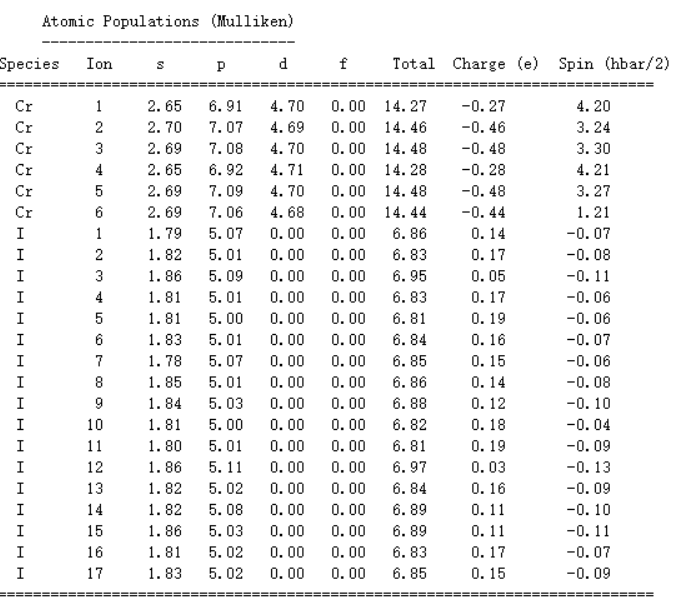
本人刚开始看缺陷态计算,有个问题想要和大家请教,缺陷形成能VS费米能级的图中,对应某种电荷态下费米能级不是确定的吗?(OUTCAR里E-Fermi),在这个图中,费米能级为什么是变化的呢?该图要表达的意思是什么呢?本人刚开始做不太明白,希望有路过的人可以耐心讲...
第一原理
- 150
- 3
借本版宝地寻有VASP使用经验的博后 [地点:北京]
wwyygg0520
诸位搞计算的虫友们好,借贵版面寻找一位有使用vasp经验的博后,欢迎大家推荐哈。地点:北京单位:某研究所(非中科院)薪水:税前23万+专业:化学/材料/物理大概方向:高压力条件下周期性体系的结构与性质模拟具体方向再根据双方兴趣协商。课题组能提供的:实验条件上大...
第一原理
- 1250
- 25

hybrid XC not allowed in non-scf calculations 求助
jackwangee
QE新手正在用HSE06,但是算NSCF时,出现hybridXCnotallowedinnon-scfcalculationsinputfile&CONTROLcalculation="nscf"max_seconds=8.6400...
第一原理
- 250
- 5

ALN的能带. 发现EF 不等于0
jackwangee
Hi,大家好我正在测试用QE做ALN的bandstructure.发现EF不等于0!但是大部分文献里是EF=0.没办法贴图。贴输入文件。&CONTROLcalculation="nscf"max_seconds=8.64000e+0...
第一原理
- 200
- 4
ALN的能带. 发现EF 不等于0
jackwangee
Hi,大家好我正在测试用QE做ALN的bandstructure.发现EF不等于0!但是大部分文献里是EF=0.没办法贴图。贴输入文件。&CONTROLcalculation="nscf"max_seconds=8.64000e+0...
第一原理
- 200
- 4
请一下各位老师:
lastnumber
我在计算cohp时,出现正交化警告,导致一直都把chargespilling降不下去,cohp手册里基本参数都试过了。不知道如何解决,有经验的就告诉我一下该如何处理。先在此谢谢各位了
第一原理
- 1950
- 39





