DMOL3 计算结果中的bind energy和homolumo可以等同于结合能电离能电子亲和能吗?
1532174329
小弟刚开始用materialsstudio,想问下各位大佬DMOL3计算结果中的bindenergy和homolumo可以等同于结合能、电离能、电子亲和能吗?
第一原理
- 300
- 6
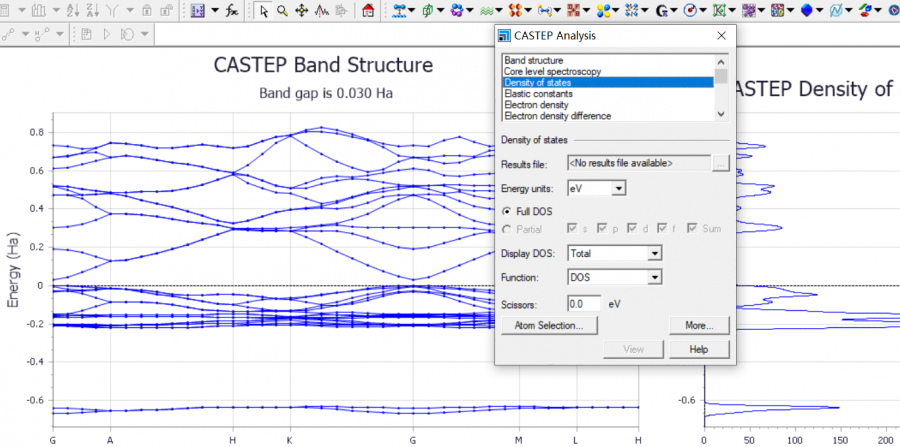

请问大家什么是lattice dynamical susceptibility

zz11ss11zz
大家好,最近看文献(PRL126,096401)发现他们用了一个叫latticedynamicalsusceptibility的量来研究CDW,但是在google查找后并没有发现太多关于该物理量的描述,想了解一下这个物理量的意义,它与latticeinstab...
第一原理
- 150
- 3
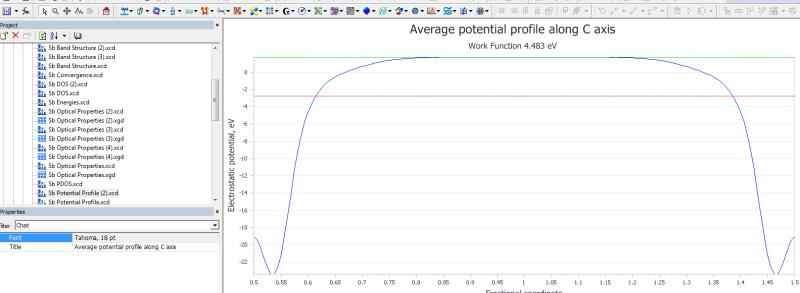


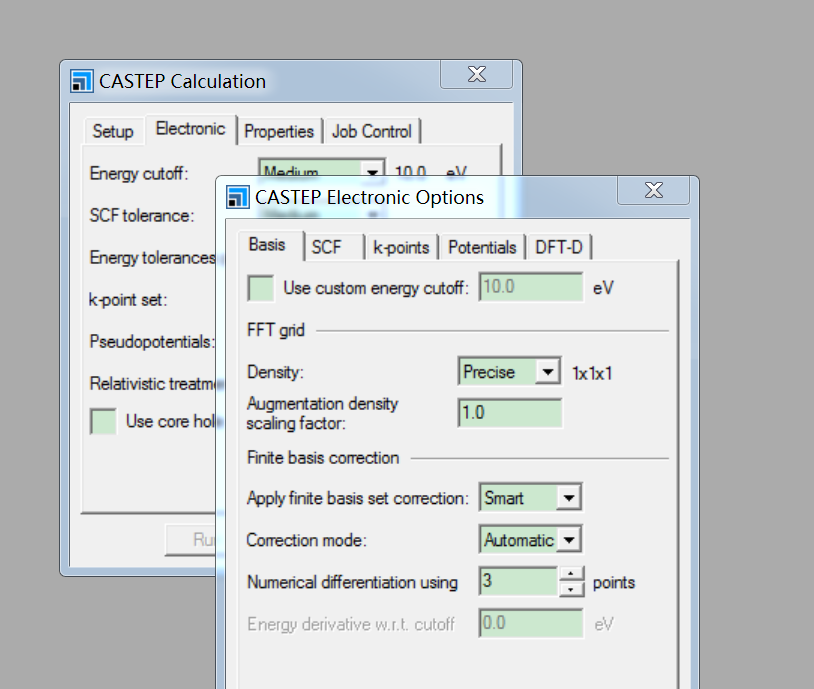





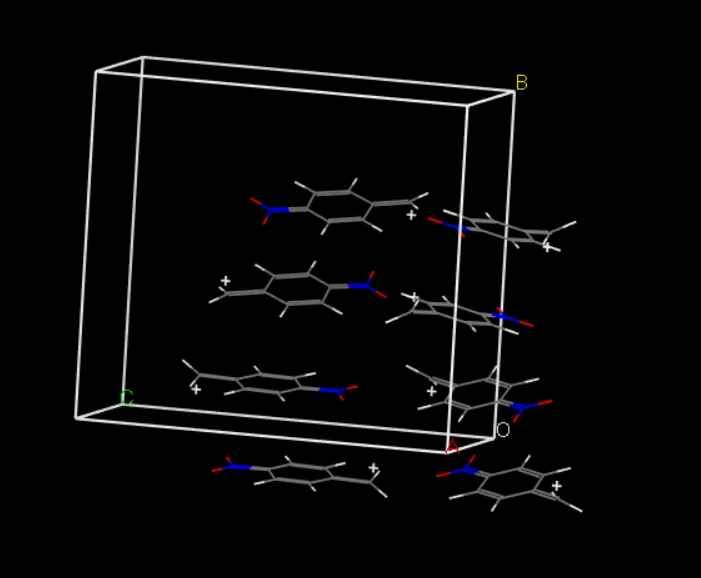
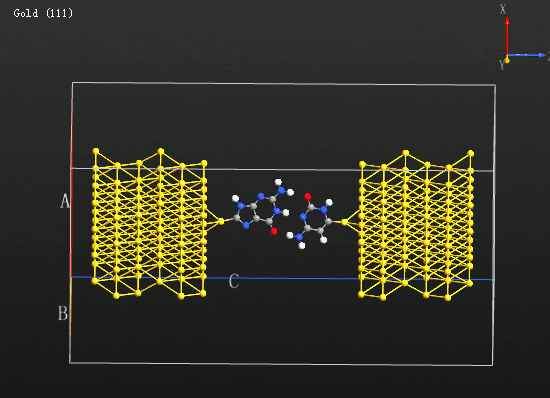
MS优化晶体结构时杂原子移动。
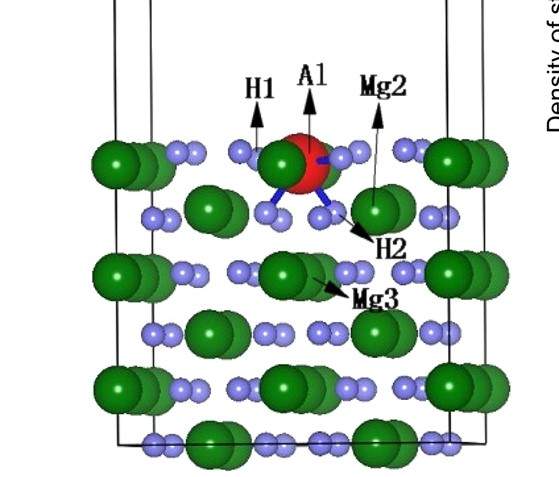

xiaopang8958
大家好,我在用MS优化含杂原子的晶体结构时,已经通过在Modify下面的constraints将其中的杂原子固定。但是在优化结构后,杂原子仍从表面移动到底部。请问各位专家...
第一原理
- 150
- 3




