vesta 显示问题

yangjian85
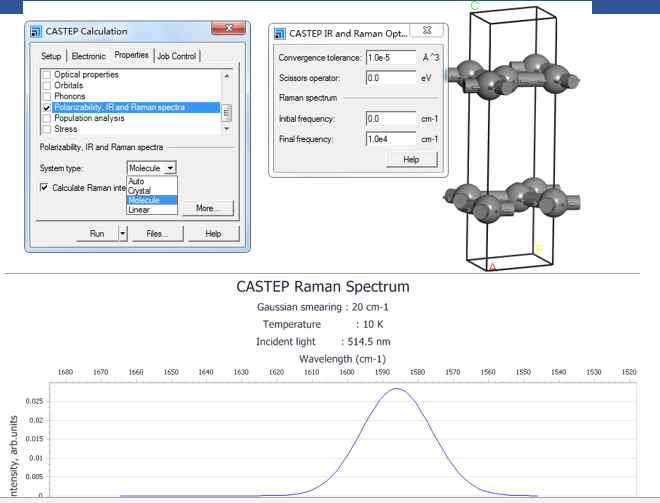
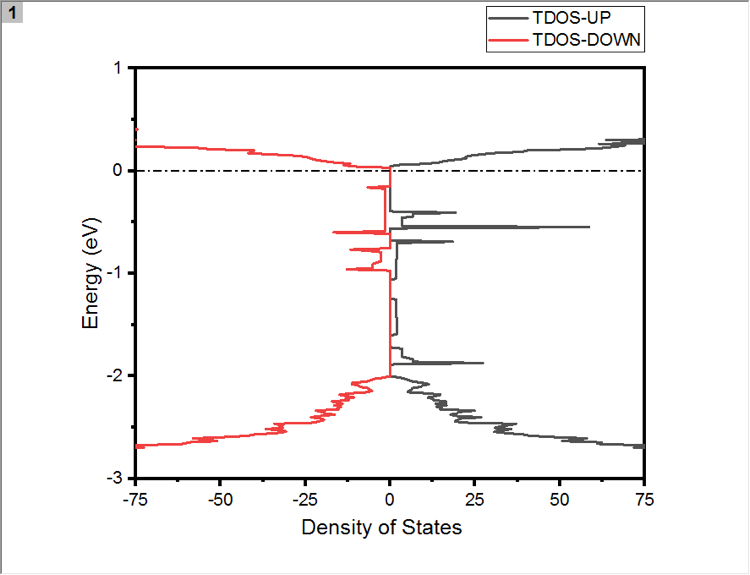
请教各位,如何设置vesta的周期性显示。我的POSCAR中只有98个原子。可是为什么我的VESTA显示的确实这个样子的?在别的电脑上就是正常显示。这是要设置什么吗?屏幕...
第一原理
- 400
- 8


澳门大学 应用物理及材料工程研究院 蔡永青课题组 博士招生(2021年秋季入学)
dielectric1
澳门大学(UniversityofMacau,UM)于1981年成立,是澳门特区唯一的一所由澳门政府全额资助的综合性多学科公立大学。经过35年的发展,澳大在教学、研究和社群服务领域均取得卓越的贡献,并已成功进入珠三角区最优秀的国际综合性大学之列,比肩香港科技大...
第一原理
- 1150
- 23

OER/ORR pH值修正

cloverliang
在使用DFT计算ORR或者OER反应的能量图和台阶图,pH是怎么影响台阶图的?𝛥𝐺PH=-kBTln[H+]=pH×kBTln10,假设pH=13,pH修正为0.77,这0.77是加在GOOH*还是加在&...
第一原理
- 650
- 13