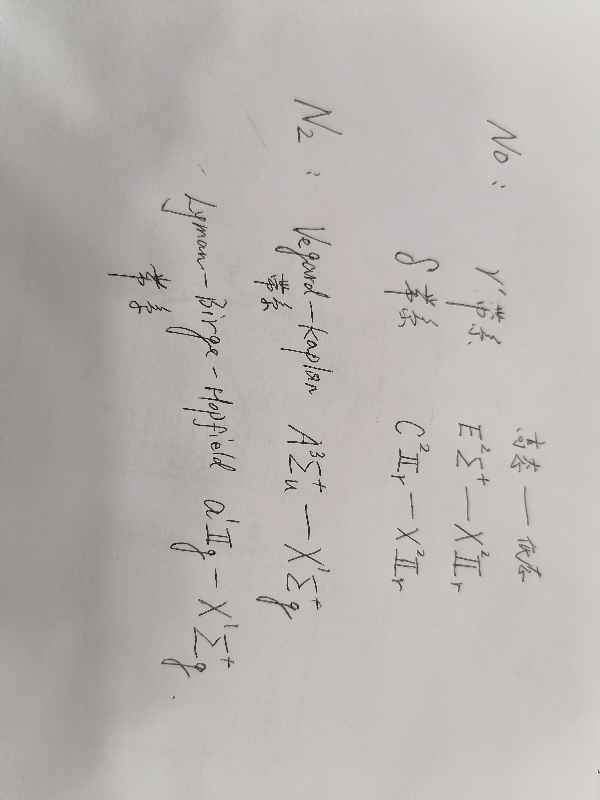



Li+与EC(碳酸乙烯酯)结合能计算
frank_xiao
本人开始学习计算,想计算Li+与不同溶剂分子的结合能比如要计算Li+与EC的结合能,直接可以从gaussianview读出charge(1)和spin(single)值,使用什么method和basisset比较好,新手还望大佬多赐教,最后能给出简单的inpu...
量子化学
- 200
- 4

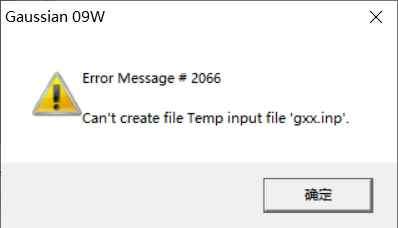


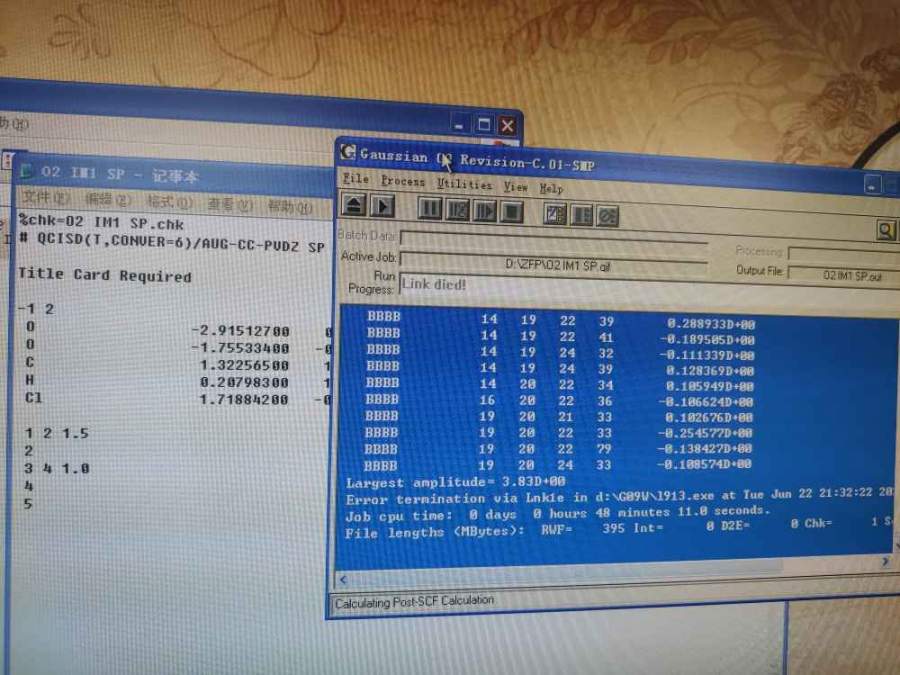



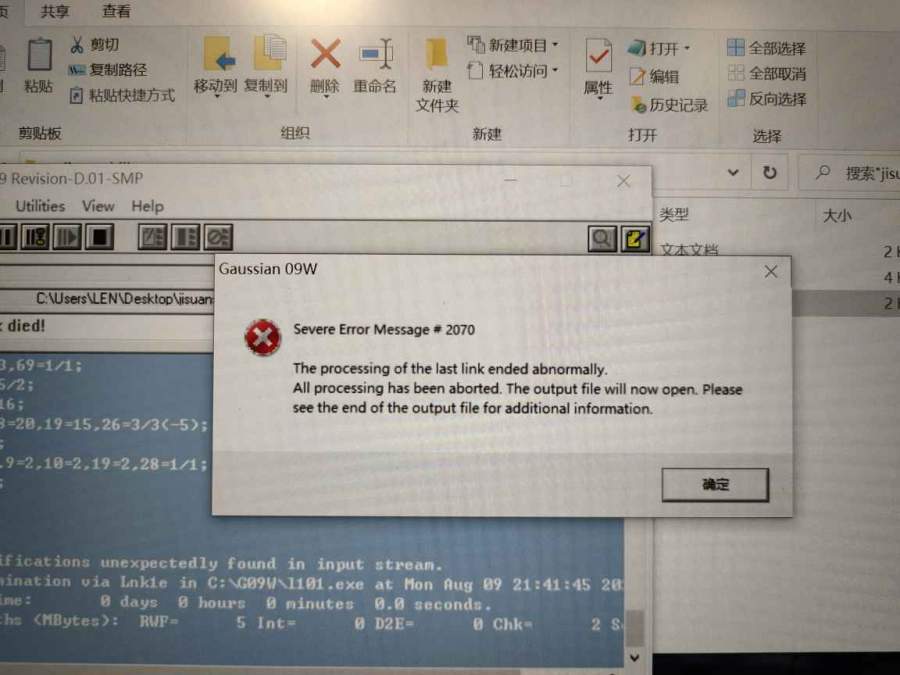



请问高斯模拟运行出错是什么原因?请求大神帮忙看看
one_piece_
EnteringLink1=G:\\GAUSSIAN\\G09W\\l1.exePID=11192.Copyright(c)1988,1990,1992,1993,1995,1998,2003,2009,2013,Gaussian,Inc.AllRightsR...
量子化学
- 250
- 5




