
高熵合金的理想强度理论计算
shaolin1120
请教各位大神,如何用VASP计算材料的拉伸强度。同过阅读文献,知道一般体心立方bcc结构沿单轴拉伸时含有两条拉伸路径(主路径PTP和次路径SOP),如沿[001]方向拉伸,对于主路径,设置初始结构为a1=a2=a3的bcc结构;对于次路径,设置初始结构为a1=...
第一原理
- 450
- 9


MS2019用castep运行报错

flyboyflyfly
MS2019用castep计算运行后除错无法继续计算下去,输出提示如下:[mpiexecDESKTOP-NUC8QLS]..\\hydra\\pm\\pmiserv\\pmiserv_cb.c(863):connectiontoproxyOathostDESK...
第一原理
- 950
- 19


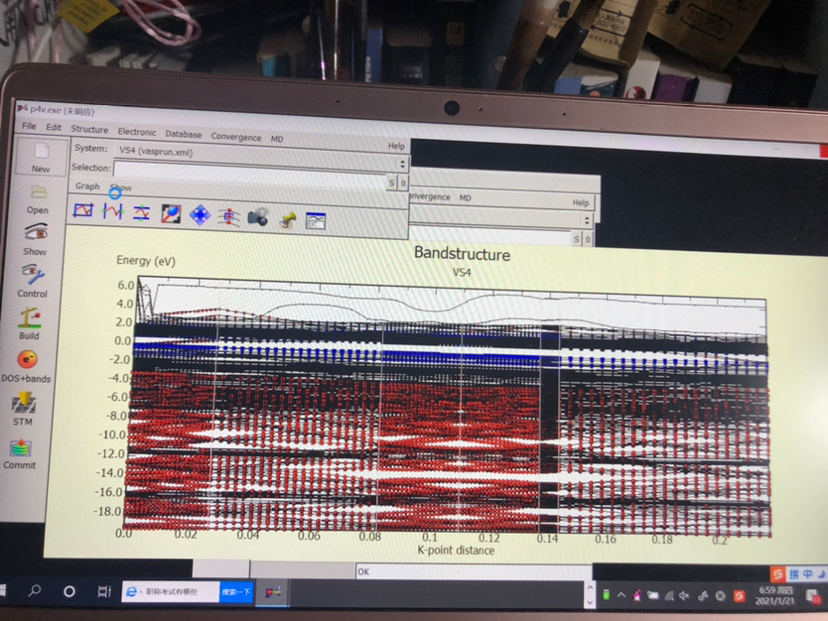


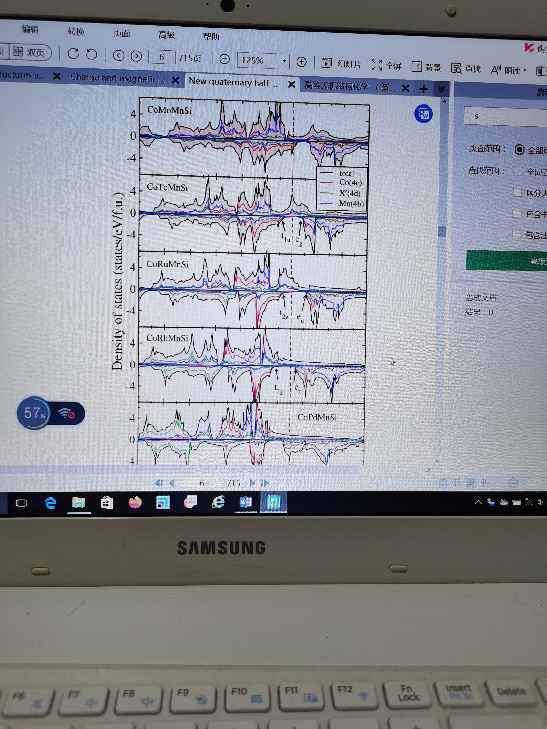

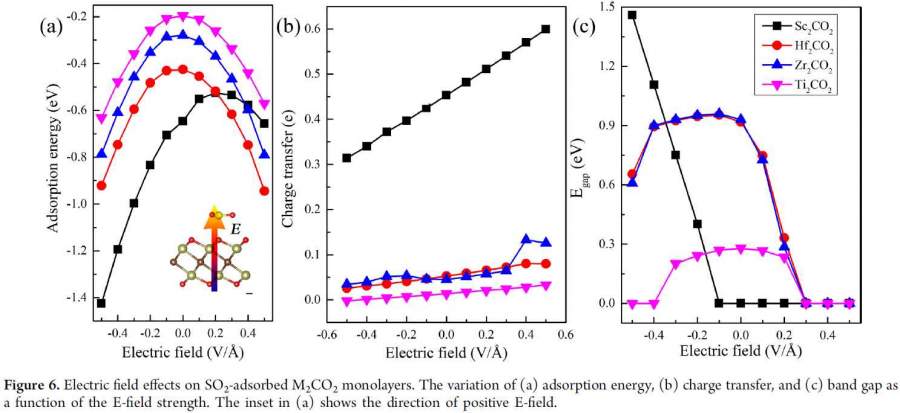



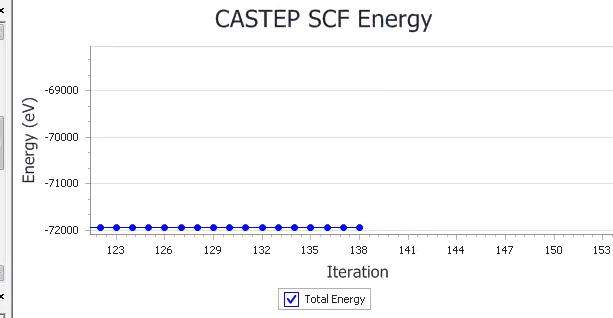
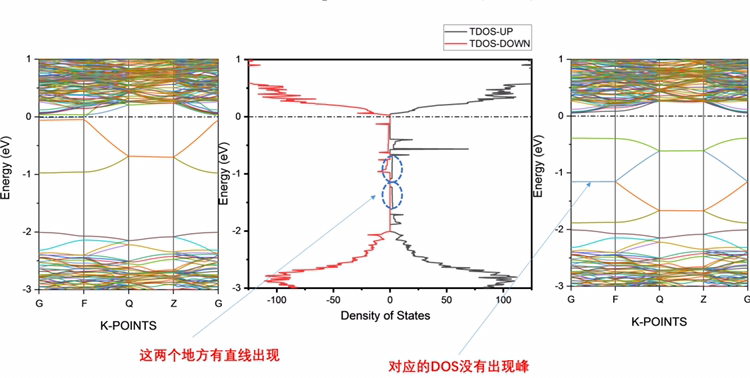
DFT计算核数和计算效率的匹配
douliguang
刚配置一台电脑,志强铂金cpu8222L两颗主频3.0G全核睿频3.4G,内存32G2666MHzDDR4RECC,一共有36核,72线程。MS计算发现核数越多计算越慢,8核计算速度比72核快好几倍,最优核数是5核。。。这样的话CPU占用率太低只有10%不到,...
第一原理
- 350
- 7