金币给你们来我这里做测试吧
naitun1173
国内专业研究微缩料,多糖解析,成分分析团队微信:18231133282(电话同号)qq:1196992037一、TEMSEMNMRICPXPSBETXRD单晶+解析核磁原位XPS球差电镜冷冻电镜等仪器极速特惠测试.二、各种大型仪器测试分析认证。为广大高校,科研...
第一原理
- 4550
- 91




费米能级和禁带宽度的问题
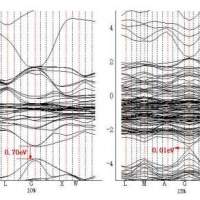

1344943084
求大佬帮忙看看,有没有可能禁带宽度在如下位置,如果按照如图中的位置的话,怎么来判断,而且按照这个数据,也和我们在实验室做出的数据相复合,但是我查了很多,大多都是按照0处费...
第一原理
- 300
- 6





澳门大学 蔡永青课题组 第一性原理计算 博士后招聘
dielectric1
研究内容:第一性原理计算下列方向之一:1)声子;2)含时密度泛函以及激发态;3)铁电以及压电性质;4)高通量计算以及人工智能。要求熟练掌握一门第一性原理计算软件如VASP,QuantumEspresso,Wien2K等等。掌握群论以及Python、Matlab...
第一原理
- 350
- 7






