
计算超精细耦合常数
1259483770
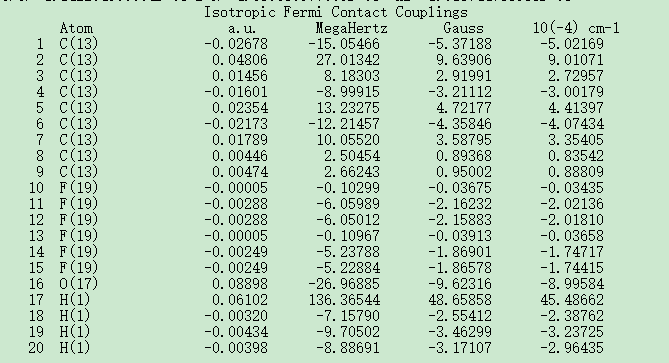
请问各位大佬,我通过Gaussian计算出来了IsotropicFermiContactCouplings,我要如何通过它来确定超精细耦合常数。请各位指教!截图.png
量子化学
- 200
- 4
计算超精细耦合常数
1259483770
请问各位大佬,我通过Gaussian计算出来了IsotropicFermiContactCouplings,我要如何通过它来确定超精细耦合常数。请各位指教!截图.png
量子化学
- 400
- 8


求助单茂金属配合物的过渡态,茂环不旋转的问题

wangyinran
寻找烯烃配位到单茂金属配合物上的过渡态,由于茂环不是对称的,环戊二烯上取代了四个甲基和一个三甲基硅基,我在寻找过渡态时,看优化过程发现茂环并没有沿着茂环中点与中心金属的连线转动,尽管这样找出来的过渡态的振动方向和IRC都符合预期。但茂环的位置会影像能量计算,怎...
量子化学
- 150
- 3
分子结构优化之前如何扫描
yangyue5678
求助各位大神,我做了个分子结构优化,有条审稿意见是Thestudiedmoleculecanpresentseveralminina,ascanonthedihedralangle4-3-10-15isnecessarybeforeproceedingtoth...
量子化学
- 400
- 8







Gaussian版权问题
wangyinran
本人在自己的服务器上安装了Gaussian16,软件从网上搜索下载。本人所在高校的超级计算机中心已购买正版G16,但只安装在超算集群上,不提供下载。在自己的服务器上算出的成果,以本人所在学校为第一单位,能否直接发表文章?
量子化学
- 150
- 3





