
寻求合作:第一性原理材料模拟及催化反应计算
chunzhenxp
课题组急寻可靠的理论计算科研工作者,合作开展研究和发表论文。本课题组主要从事电化学催化的机理研究,侧重材料的原位谱学表征,对于一些材料的吸附脱附性能,bandstructure等都需要开展理论计算方面的合作。合作对象可以是博后,研究员,或者课题组的PI。要求理...
第一原理
- 250
- 5


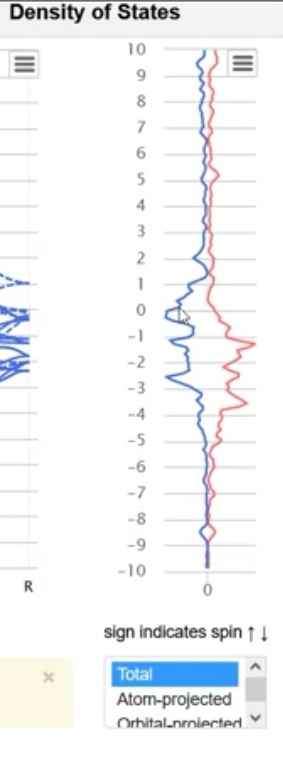
materials project如何提取DOS
fgmsyutong
'https://www.materialsproject.org/materials/mp-1018101/'materialsprojects,如何提取该合金某一点的...
第一原理
- 5000
- 100

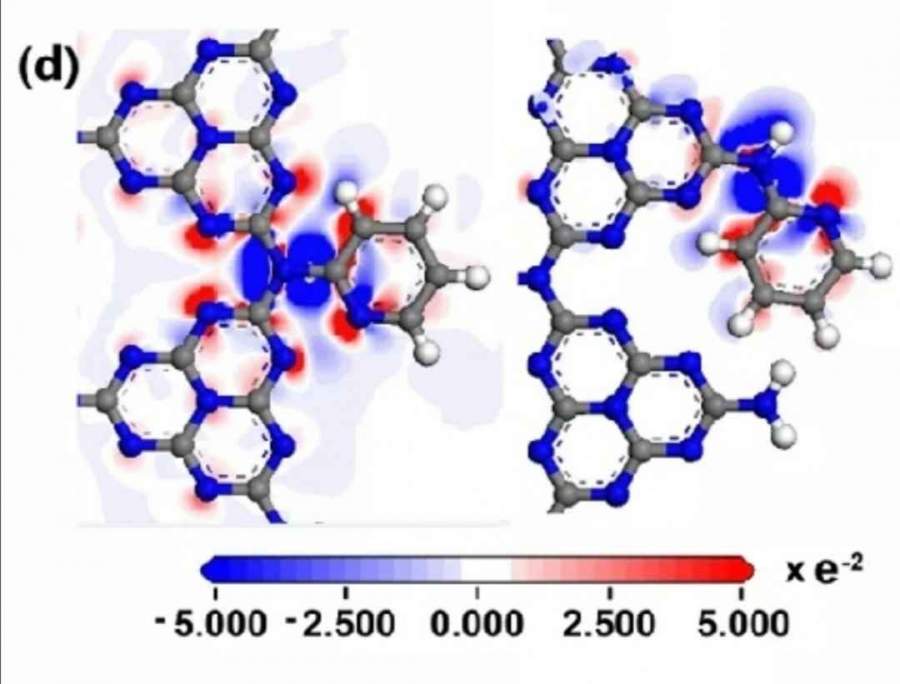

HSE06-band计算不进行迭代

1677852360
我在进行HSE06进行能带计算时,先用vaspkit生成所用的k点,进行了pbe-scf,然后用原来的kpoints、poscar、potcar以及自洽产生的waveca...
第一原理
- 1050
- 21
求助大佬CL-NEB计算问题
lingfaling
计算一步收敛,受力确为0。但这也只是针对某些体系,有的体系能够正常计算,并得出结果。检查过中间的结构,没得问题。主要参数设置:NSW=200#numberofstepsforionicupdIBRION=3#ionicrelaxation0=MD1=quasi...
第一原理
- 200
- 4
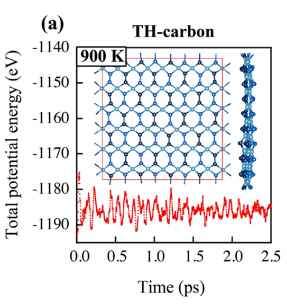
高对称点不连续,计算出来的能带是断层的,要怎么计算。
1677852360
我想问下,我在用PBE计算能带结构时,发现用vaspkit生成的高对称点是不连续的,因此算出来的能带是有轻微断层的,不相连,这种情况要怎么解决呢??希望有大神可以给出建议。
第一原理
- 500
- 10





