



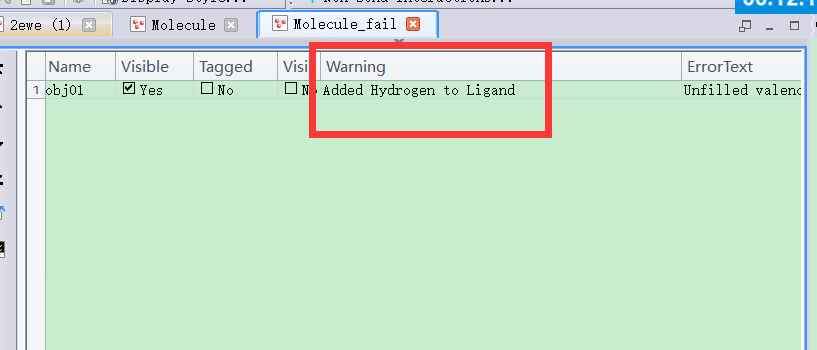
Ds对接时显示配体有问题

17363392860
CDOCKER对接时,有时error就是Addedhydrogentoligand,对配体进行prepareligand以后对接显示error:Norefinedpose...
分子模拟
- 500
- 10



建立qsar模型,为什么训练集化合物数量不应过多?
SunRuikang
《计算机辅助药物设计》指明,“用于构建方程的训练集最好不要超过50个化合物”。个人认为,训练集化合物数量越多越好,像是过拟合等问题,应该从算法找原因,不是训练集过多的问题。为什么qsar的训练集不能过多?谢谢同好们!
分子模拟
- 250
- 5
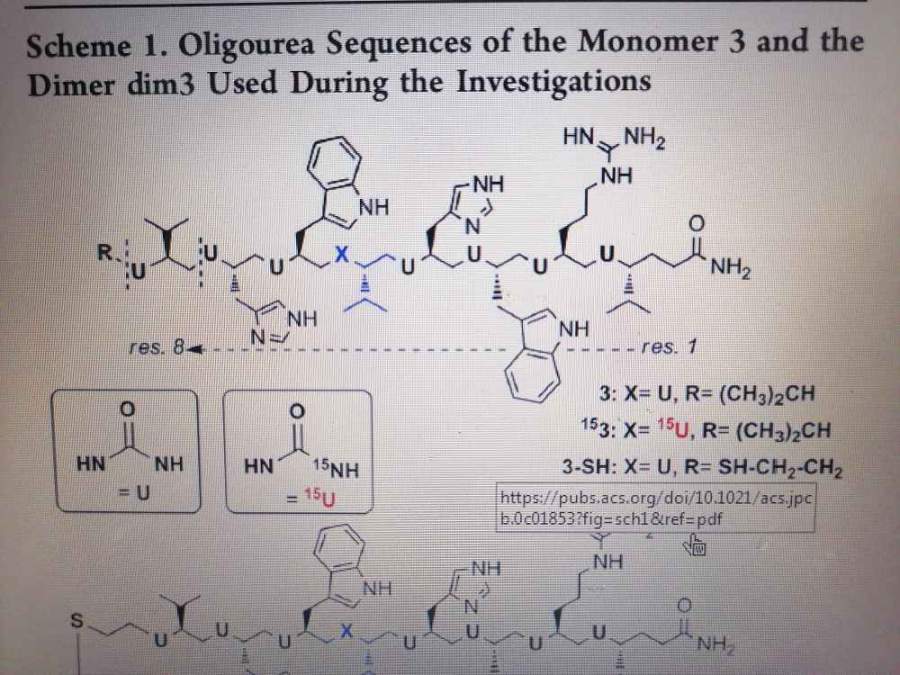





COMPASS力场找不到Mg金属,只有离子态和氧化态,求助!!
nico910902
大家好,最近在做镁金属对有机物的吸附,在做分子动力学的过程中发现用COMPASS/COMPASSII力场竟然找不到Mg,只有Mg2+和MgO.但是查了官方数据后发现COMPASS力场里是有Mg金属的。请问这是什么情况呀?我用的是MaterialStudio20...
分子模拟
- 250
- 5
gromacs计算速度
Yolanda9288
实验室有两台工作站,cpu相同,均没有gpu,只有内存差别,一个256g,一个384g,但是我在两台机器上计算一样的模型速度差别很大,内存高的快很多,但我看计算时,内存占用率并不高,cpu占用率都是一样的,是什么原因呢?慢的那台是不是安装gromacs的时候哪...
分子模拟
- 300
- 6



