计算中自旋多重度的选择的疑惑 新手求助
大家好,之前在计算中性体系时电荷和自旋多重度分别设置值为0和1,都能够顺利计算。
最近在计算一个过渡金属配位体系(如图1)时发现自己画好的结构生成的gjf文件里电荷和自旋多重度默认值分别为0和2,
把它改为0和1后计算提示体系497个电子和自旋多重度1不匹配(如图2),所以又改回了0和2,最后计算的结果发现电子排布的示意图里成了2列(如图3),
不明白的地方是:1.电荷为0,自旋多重度为2意味着什么?
2.电子排布示意图呈2列出现,怎样分析这种结果?对后续的计算(如TD、nto)有什么影响?
感谢大家的帮助,谢谢!
附输入文件:
%chk=1.chk
%mem=32GB
%nprocshared=32
# opt b3lyp/genecp scf=maxcyc=130
s0 opt
0 2
。。。(笛卡尔坐标)
C H N 0
6-31G(d,p)
****
Cu 0
LanL2dz
****
Cu 0
LanL2dz

1.jpg

2.JPG

3.jpg![]() 返回小木虫查看更多
返回小木虫查看更多
今日热帖






补充:在结构里添加两个硝酸根离子后依然提示奇数电子和0.1不匹配,无法计算。
关于电荷和自旋多重度的判断可以见我这个教程的1.2节 https://muchong.com/t-14242735-1,截图如下

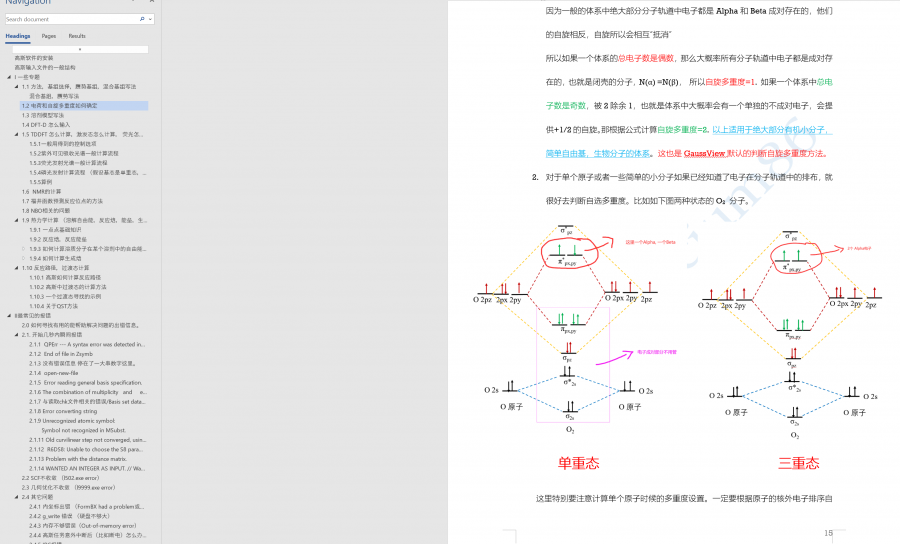

自旋.png
自旋2.png
自旋3.png
,
谢谢版主回复!
我已经将您的这篇文献打印成册,正在拜读中,您指出的这几页我也读过,仍然不太懂自旋多重度是2的物理或者化学意义,请问需要补充哪方面的知识呢?
自旋多重度,本身就是体系的电子的总自旋量子数决定的。至于电子自旋量子数有什么物理意义,那应该取看量子化学的教材。高斯需要输入文件中包含这个信息,主要是为了大致确定电子在轨道里如何排布。有几个alpha电子几个beta电子, 这个会影响解SCF的过程。这部分你得看基础的计算化学教材。 另外在磁场中,二重态分子会裂分成两个能级 三重态列分成三个能级 等等 这也是这个叫法的由来。
谢谢版主,继续学习