
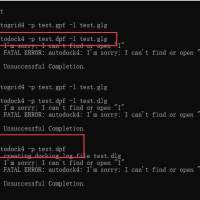






Ds对接时显示配体有问题

17363392860
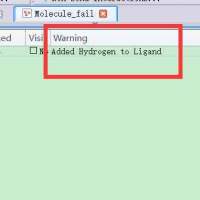
CDOCKER对接时,有时error就是Addedhydrogentoligand,对配体进行prepareligand以后对接显示error:Norefinedpose...
分子模拟
- 500
- 10

招收博后~分子模拟~风味研究~工作地点北上广~自由度高
naughtydog
广州大学昆虫食品创新团队与上海交通大学食品风味创新团队联合招收博士后'https://mp.weixin.qq.com/s/BeftK_rG1M5f1ZqHtHx8Tw'一、招聘生物、化学、生理、生化、传感、神经、分子模拟、食品风味相关学科背景的海内外博士。二...
分子模拟
- 1150
- 23





建立qsar模型,为什么训练集化合物数量不应过多?
SunRuikang
《计算机辅助药物设计》指明,“用于构建方程的训练集最好不要超过50个化合物”。个人认为,训练集化合物数量越多越好,像是过拟合等问题,应该从算法找原因,不是训练集过多的问题。为什么qsar的训练集不能过多?谢谢同好们!
分子模拟
- 250
- 5





